SPAGxEmixCCT-local
SPAGxEmixCCT-local is a scalable and accurate G×E analysis framework that allows tests for ancestry-specific effects by incorporating local ancestry in multi-way admixed populations.
Introduction of SPAGxEmixCCT-local
SPAGxEmixCCT-local extends SPAGxEmixCCT by integrating local ancestry information to enhance the precision of ancestry-specific G×E effect testing and statistical powers. Furthermore, we introduce SPAGxEmixCCT-local-global, which combines p-values from both SPAGxEmixCCT and SPAGxEmixCCT-local, offering an optimal unified approach for various cross-ancestry genetic architectures. As with SPAGxEmixCCT, SPAGxEmixCCT-local involves two main steps:
-
Step 1: SPAGxEmixCCT-local fits a genotype-independent (covariates-only) model and calculates the model residuals. Details are described in Step 1 of SPAGxECCT.
-
Step 2: SPAGxEmixCCT-local identifies genetic variants with marginal G×E effects on the trait of interest. First, SPAGxEmixCCT-local estimates the ancestry-specific allele frequencies for the variants. Next, SPAGxEmixCCT-local evaluates ancestry-specific marginal genetic effects using ancestry-specific score statistics. If an ancestry-specific marginal genetic effect is not significant, we use ancestry-specific test statistic SG×E(mix) to characterize the ancestry-specific marginal G×E effect. If significant, SG×E(mix) is updated to ancestry-specific genotype-adjusted test statistics. The hybrid strategy to balance computational efficiency and accuracy follows SPAGxECCT.
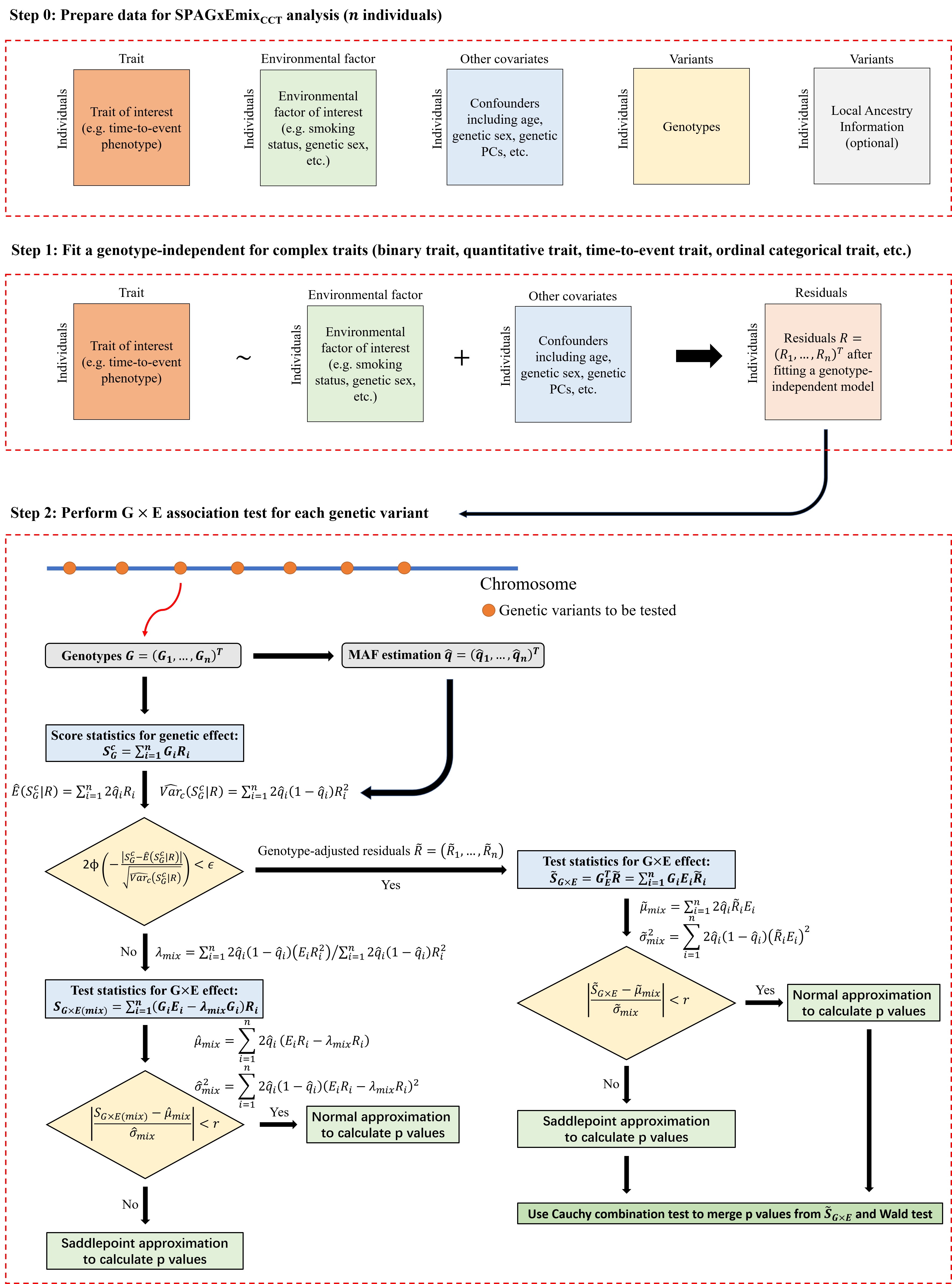
Compared to conventional standard statistical testing methods that account for local ancestry, SPAGxEmixCCT-local offers much greater computational efficiency.
- SPAGxEmixCCT-local leverages local ancestry information to estimate the distribution of ancestry-specific genotypes and the null distribution of test statistics.
- For most tests (ancestry-specific genetic main effect p-values > 0.001) in a genome-wide G×E analysis, SPAGxEmixCCT-local utilizes residuals from a genotype-independent model (fitted only once across the genome-wide analysis) to construct test statistics.
- Conventional methods have to incorporate local ancestry as covariates and fit a separate model for each variant, which is computationally burdensome for genome-wide analyses.
Quick start-up examples
The following example illustrates how to use SPAGxEmixCCT-local to analyze a binary trait.
Step 1. Read in data and fit a genotype-independent model
library(SPAGxECCT)
# load in phenotype, genotype, and local ancestry counts for two ancestries
data("Pheno.mtx") # phenotype data
data("Geno.mtx") # genotype data
data("Geno.mtx.ance1") # ancestry-specific genotype data for ancestry 1
data("Geno.mtx.ance2") # ancestry-specific genotype data for ancestry 2
data("haplo.mtx.ance1") # local ancestry counts for ancestry 1
data("haplo.mtx.ance2") # local ancestry counts for ancestry 2
Cova.mtx = Pheno.mtx[,c("PC1", "PC2", "PC3", "PC4", "Cov1", "Cov2")] # Covariate matrix excluding environmental factor
E = Pheno.mtx$E # environmental factor
### Cova.haplo.mtx.list
Cova.haplo.mtx.list = list(haplo.mtx.ance1 = haplo.mtx.ance1,
haplo.mtx.ance2 = haplo.mtx.ance2) # local ancestry counts of ancestry 1 and 2
### fit a genotype-independent model for the SPAGxEmix_CCT_local analysis
resid = SPA_G_Get_Resid(traits = "binary",
y ~ Cov1 + Cov2 + E + PC1 + PC2 + PC3 + PC4,family=binomial(link="logit"),
data = Pheno.mtx,
pIDs = Pheno.mtx$IID,
gIDs = rownames(Geno.mtx))
Step 2. Conduct a marker-level association study
### calculate p values for ancestry-specific GxE effects of ancestry 1
binary_res_ance1 = SPAGxEmixCCT_localance(traits = "binary",
Geno.mtx = Geno.mtx.ance1,
R = resid,
haplo.mtx = haplo.mtx.ance1,
E = E,
Phen.mtx = Pheno.mtx,
Cova.mtx = Cova.mtx,
Cova.haplo.mtx.list = Cova.haplo.mtx.list)
colnames(binary_res_ance1) = c("Marker", "MAF.ance1","missing.rate.ance1",
"Pvalue.spaGxE.ance1","Pvalue.spaGxE.Wald.ance1", "Pvalue.spaGxE.CCT.Wald.ance1",
"Pvalue.normGxE.ance1", "Pvalue.betaG.ance1",
"Stat.betaG.ance1","Var.betaG.ance1","z.betaG.ance1")
### calculate p values for ancestry-specific GxE effects of ancestry 2
binary_res_ance2 = SPAGxEmixCCT_localance(traits = "binary",
Geno.mtx = Geno.mtx.ance2,
R = resid,
haplo.mtx = haplo.mtx.ance2,
E = E,
Phen.mtx = Pheno.mtx,
Cova.mtx = Cova.mtx,
Cova.haplo.mtx.list = Cova.haplo.mtx.list)
colnames(binary_res_ance2) = c("Marker", "MAF.ance2","missing.rate.ance2",
"Pvalue.spaGxE.ance2","Pvalue.spaGxE.Wald.ance2", "Pvalue.spaGxE.CCT.Wald.ance2",
"Pvalue.normGxE.ance2", "Pvalue.betaG.ance2",
"Stat.betaG.ance2","Var.betaG.ance2","z.betaG.ance2")
### merge data frame
binary.res = merge(binary_res_ance1, binary_res_ance2)
# we recommand using column of 'p.value.spaGxE.CCT.Wald.index.ance' to associate genotype with phenotypes
head(binary.res)
Citation
- A scalable and accurate framework for large-scale genome-wide gene-environment interaction analysis and its application to time-to-event and ordinal categorical traits (to be updated).